Cell injury
4 years ago 12093
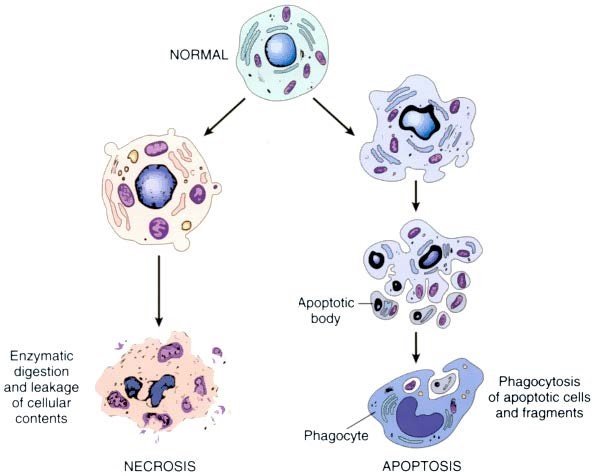
Cell injury:
Definition of cell injury: If the limits of adaptive responses are exceeded, or if cells are exposed to injurious agents or stress, deprived of essential nutrients, or become compromised by mutations that affect essential cellular constituents, a sequence of events follows that is termed cell injury. (Ref Robbin’s pathology, Page 35)
Type of cell injury:
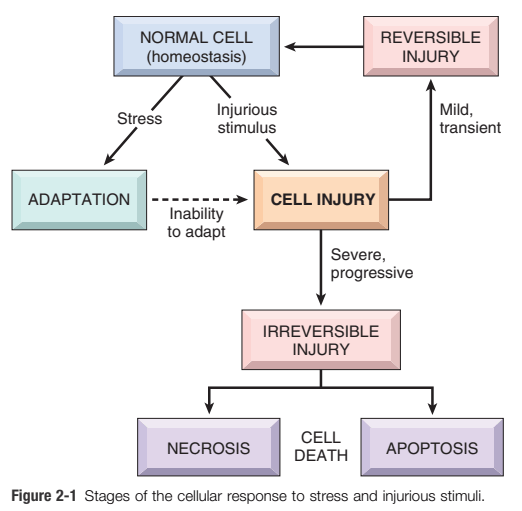
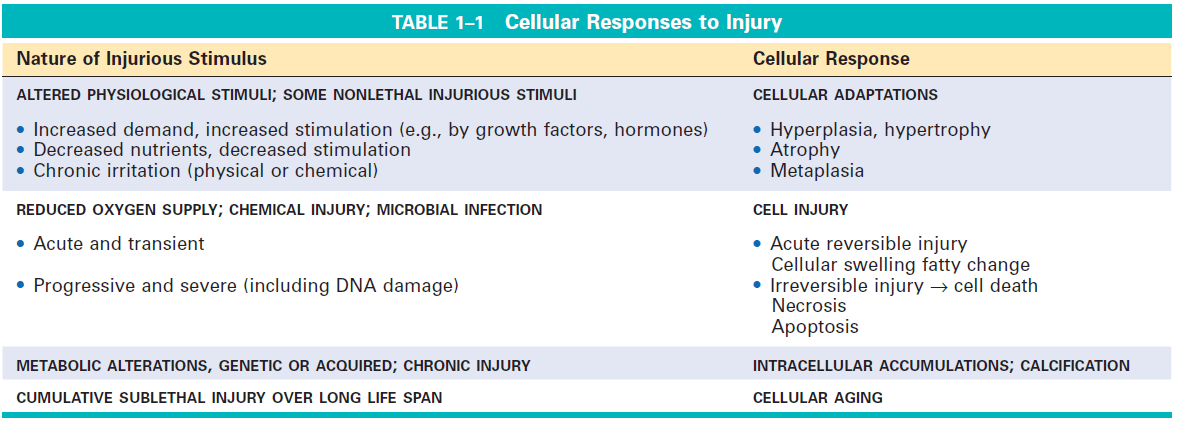
Morphological Types of cell injury:
| Reversible cell injury | Irreversible cell injury |
| 1) Cellular swelling (first manifestation of almost all forms of cell injury) 2) Fatty change |
1) Necrosis 2) Apoptosis 3) Necroptosis 4) Pyroptosis 5) Ferroptosis |
Causes of cell injury:
1. Oxygen deprivation
Hypoxia: Deficiency of oxygen, causes cell injury by reducing aerobic oxidative respiration.
2. Physical agents:
Mechanical trauma, extremes of temperature (burns and deep cold), sudden changes in atmospheric pressure, radiation and electric shock.
3. Chemical agents and drugs:
4. Infectious agents: Virus, Bacteria, fungus, Rickettsia, parasites.
5. Immunologic reaction: Anaphylactic reaction and Autoimmune diseases
6. Genetic abnormalities: Congenital malformations, enzyme deficiency (storage diseases).
7. Nutritional abnormalities: Protein-energy malnutrition, obesity, atherosclerosis
Important targets of injurious stimuli
Role of ATP depletion
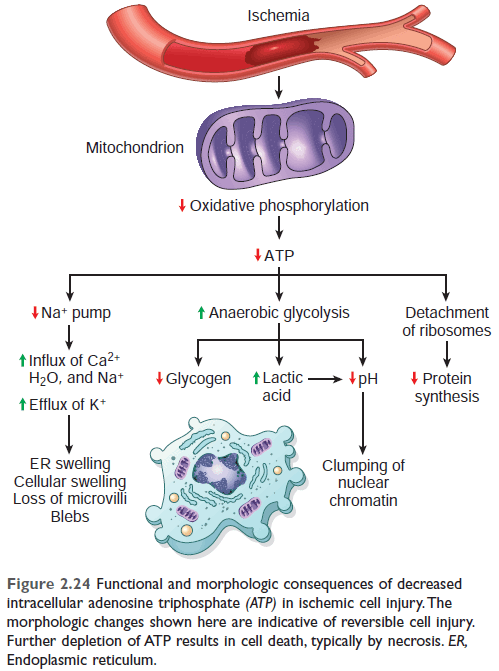
Figure: Functional and morphologic consequences of reversible cell injury in ischemia (ATP depletion). Ref Robbin's 10th edition (Page 56)
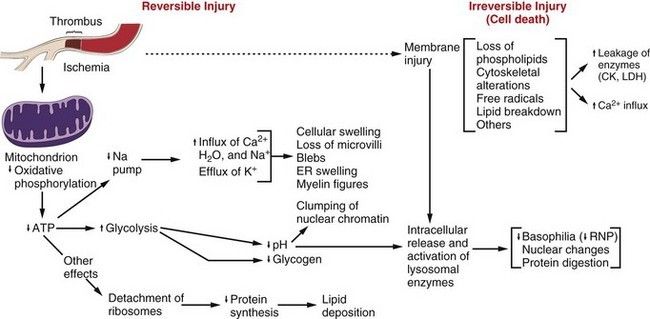
Figure: Sequence of events in reversible and irreversible cell injury (Robbin's 7th Edition)
Role of Mitochondria:
Cause of mitochondrial damage:
Consequences of mitochondrial damage:
There are two major consequences of mitochondrial damage:
1. Mitochondrial damage often results in the formation of a high-conductance channel in the mitochondrial membrane, called the mitochondrial permeability transition pore.
The opening of this conductance channel leads to the loss of mitochondrial membrane potential, resulting in failure of oxidative phosphorylation and progressive depletion of ATP, culminating in necrosis of the cell.
2. Increased permeability of the outer mitochondrial membrane may result in leakage of cytochrome c and proteins into the cytosol that indirectly activates apoptosis-inducing enzymes called caspases.
(Induce Apoptotic cell death: Release of Cytochrome C and protein into cytoplasm → Activate apoptosis by caspases.)
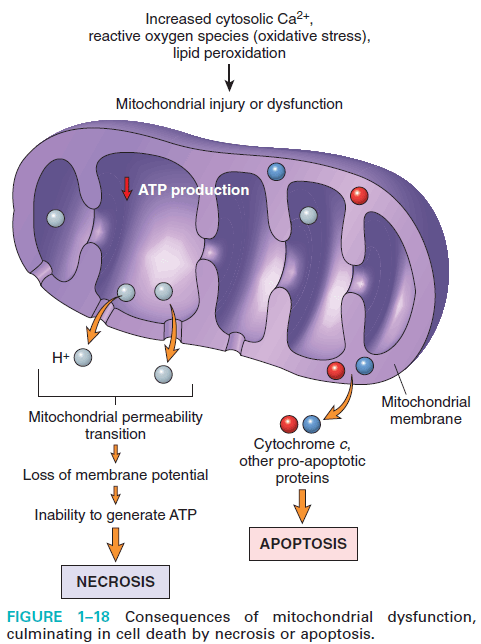
Figure: Mitochondrial dysfunction by cell injury (Robbin's 9th Edition)
The influx of calcium and loss of calcium homeostasis
Free calcium concentration is
Store site: Most intracellular calcium is sequestered in mitochondria and the ER.
Role of calcium:
A. Ischemia and certain toxins cause an increase in cytosolic calcium concentration, initially because of the release of Ca2+ from intracellular stores, and later resulting from increased influx across the plasma membrane.
B. Increased intracellular Ca2+ causes cell injury by several mechanisms.
1) Accumulation of Ca2+ in mitochondria results in the opening of the mitochondrial permeability transition pore and failure of ATP generation.
2) Increased cytosolic Ca2+ activates a number of enzymes, including
3) Induce apoptosis, by direct activation of caspases and by increasing mitochondrial permeability.
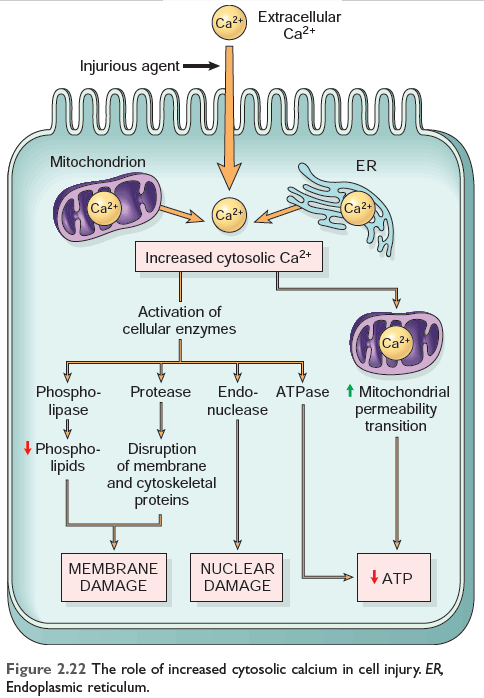
FIGURE: The role of increased cytosolic calcium in cell injury Robbin's 10th Edition
Role of Free radical:
Free radicals:
Free radicals are chemical species that have a single unpaired electron in an outer orbit.
Oxidative stress: Accumulation of oxygen-derived free radicals.
Free radical-mediated damage includes:
Generation of free radicals:
A. The reduction-oxidation reactions occur during normal metabolic processes.
This leads to the generation of Superoxide anion (O2-), Hydrogen peroxide (H2O2), and Hydroxyl ions (OH).
B. Absorption of radiant energy (e.g., ultraviolet light, x-rays): hydrolyze water into hydroxyl (OH) and hydrogen (H) free radicals.
C. Rapid bursts of ROS are produced in activated leukocytes during inflammation.
D. Enzymatic metabolism of exogenous chemicals or drugs (e.g., carbon tetrachloride CCI 4 can generate CCI3).
Transition metals:
(Iron and copper) donate or accept free electrons during intracellular reactions and catalyze free radical formation, as in the Fenton reaction (H2O2 + Fe2+ —> Fe3+ + OH + OH -).
Nitric oxide (NO), acts as a free radical and can also be converted to highly reactive peroxynitrite anion (ONOO-) as well as NO2 and NO3-.
Removal of free radicals:
1) Spontaneous decay:
For example, superoxide decay spontaneously into O2 and H2O2 in the presence of water.
2) Inactivation of free radicals:
A. Enzymatic mechanism of Inactivation of free radicals:
B. Non-enzymatic mechanism Inactivation of free radicals:
I. Antioxidant:
Example: Lipid soluble Vitamin A, E, C, and glutathione.
Mechanism: Block initiation of free radical formation or inactivate free radicals.
II. Transport protein:
Examples: Transferrin, ferritin, lactoferrin, and ceruloplasmin.
Mechanism: Minimize free radical formation by binding with iron and copper.
Pathological effects of Free Radicals:
Mechanism of free radical injury:
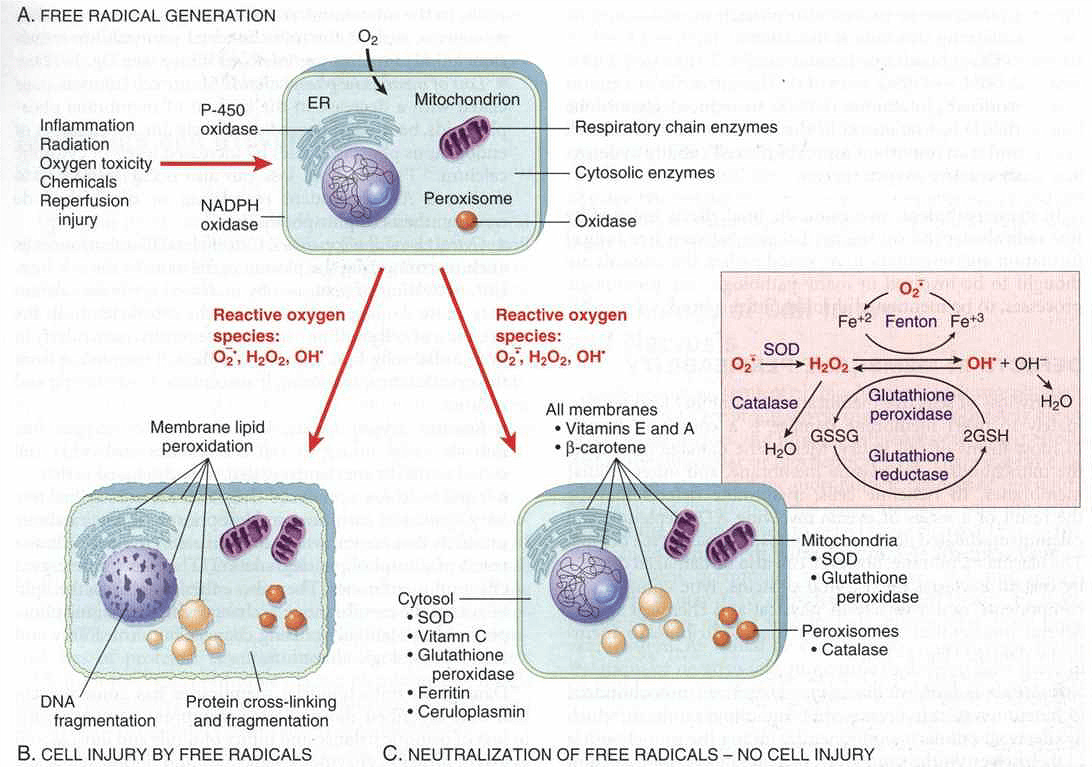
Figure: Free radical generation and inactivation (Robbins 2005 7th Edition)
Cause of Membrane damage:
Biochemical mechanism of membrane damage:
Consequences of membrane damage:
Mitochondrial membrane damage - discussed earlier.
Plasma membrane damage - Plasma membrane damage results in loss of osmotic balance and influx of fluids and ions, as well as loss of cellular contents.
Injury to lysosomal membranes - results in leakage of their enzymes into the cytoplasm and digestion of cell components (necrosis).
Damage to DNA and Proteins: Consequence
Accumulation of damaged DNA and misfolded proteins triggers apoptosis.
Conclusion:
Reversible vs irreversible Injury
1) ‘The point of no return’ at which the damage becomes irreversible, is still largely undefined.
2) Two phenomena consistently characterize irreversibility—the inability to reverse mitochondrial dysfunction and profound disturbances in membrane function.
3) Provides a means of detecting tissue-specific cellular injury and necrosis using blood serum samples (due to leakage of intracellular proteins through the damaged cell membrane and ultimately into the circulation)
Clinicopathologic correlation
Ischaemic – Reperfusion Injury
Restoration of blood flow to ischemic tissues can promote recovery of cells if they are reversibly injured, but can also paradoxically exacerbate the injury and cause cell death.
Clinical importance: Contributes to tissue damage during MI and cerebral infarction and following therapies to restore blood flow.
What causes reperfusion injury:
by new damaging process are set in motion during reperfusion, causing death of cells that might have recovered otherwise.
Mechanism:
Definition: An abnormal accumulation of triglyceride within parenchymal cells is called steatosis (fatty change).
Common sites of steatosis: Most common - Liver, also heart, muscle, and kidney.
The predisposing factor of steatosis:
Mechanism of steatosis (fatty changes):
1. Excess entry of free fatty acids from ingested food or adipose tissue. Eg. Starvation, corticosteroid.
2. Increased fatty acid synthesis from acetate.
3. Decreased fatty acid oxidation.
Both excess entry of free fatty acid and decreased fatty acid oxidation results in increased esterification of fatty acid to triglyceride.
4. Increase esterification of FA to triglyceride due to increased ?-glycerophosphate. Eg-alcoholism.
5. Decreased apoprotein synthesis.
6. Impaired lipoprotein secretion from the liver.
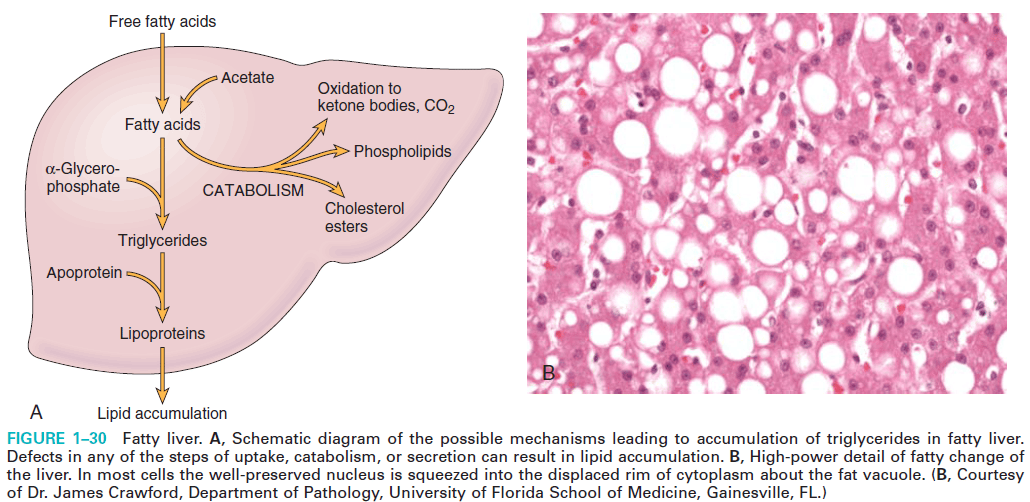
Figure: Mechanism of fatty change (Robbin's 9th Edition)
Microscopic examination:
Initially formation of small vacuoles in the cytoplasm around the nucleus.
As the process progresses, the vacuoles coalesce, creating cleared spaces that displace the nucleus to the periphery of the cell.
Types of vacuoles:
The vacuole can be differentiated by the special stain.
Differentiation of the vacuole:
Special stain for fat:
Special stain for Glycogen:
Morphology of cell injury
Morphology of Reversible cell injury:
Ultrastructural changes of reversible cell injury:
Irreversible cell injury:
Definition: The morphologic appearance of necrosis, as well as necroptosis, is the result of the denaturation of intracellular proteins and enzymatic digestion of the lethally injured cell.
Source of enzyme: The enzymes are derived either -
Morphology of necrosis:
Karyolysis: The basophilia of the chromatin may fade, reflects the loss of DNA because of enzymatic degradation by endonucleases.
Pyknosis (2nd pattern), is characterized by nuclear shrinkage and increased basophilia. Here the DNA apparently condenses into a solid, shrunken basophilic mass.
This pattern is also seen in apoptosis.
Karyorrhexis (3rd pattern), the pyknotic or partially pyknotic nucleus undergoes fragmentation.
Within 1-2 days the nucleus totally disappears.
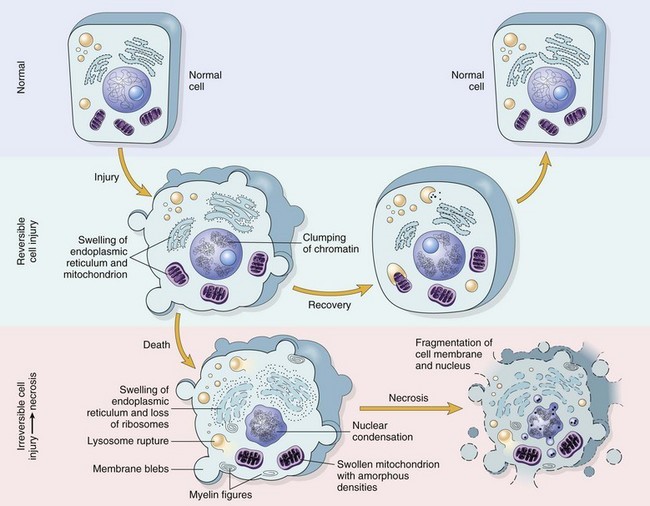
Figure: Schematic representation of a normal cell and the changes in reversible and irreversible cell injury.
Reversible injury is characterized by generalized swelling of the cell and its organelles; blebbing of the plasma membrane; detachment of ribosomes from the endoplasmic reticulum; and clumping of nuclear chromatin. Transition to irreversible injury is characterized by increasing swelling of the cell; swelling and disruption of lysosomes; the presence of large amorphous densities in swollen mitochondria; disruption of cellular membranes; and profound nuclear changes.
The latter include nuclear condensation (pyknosis), followed by fragmentation (karyorrhexis) and dissolution of the nucleus (karyolysis).
Laminated structures (myelin figures) derived from damaged membranes of organelles and the plasma membrane first appear during the reversible stage and become more pronounced in irreversibly damaged cells. The mechanisms underlying these changes are discussed in the text that follows.
From: Robbins © 2005 Elsevier
Type of Necrosis:
| Basic type | Special type: |
|
1. Coagulative necrosis
2. Liquefactive necrosis |
1. Gangrenous necrosis 2. Caseous necrosis 3. Fat necrosis 4. Fibrinoid necrosis |
Coagulative necrosis is a form of necrosis in which the architecture of dead tissues is preserved.
Liquefactive necrosis: is characterized by the digestion of the dead cells, resulting in the transformation of the tissue into a liquid viscous mass.
The term “caseous” (cheeselike) is derived from the friable white appearance of the area of necrosis. It is the distinctive form of coagulative necrosis.
Example: tuberculous infection.
Grossly: Necrotic area appears cheesy white.
On microscopic examination:
the necrotic area appears as a collection of fragmented or lysed cells and amorphous granular debris enclosed within a distinctive inflammatory border;
fat necrosis refers to focal areas of fat destruction, typically resulting from the release of activated pancreatic lipases into the substance of the pancreas and the peritoneal cavity. These occur in acute pancreatitis.
Pathogenesis (Enzymatic):
Morphology of Fat necrosis:
Clinical consequence:
The earliest histologic evidence of myocardial necrosis does not become apparent until 4 to 12 hours later. However, because of the loss of plasma membrane integrity, cardiac-specific enzymes and proteins are rapidly released from necrotic muscle and can be detected in the blood as early as 2 hours after myocardial cell necrosis.
Apoptosis is a form of cell death, designed to eliminate unwanted host cells through the activation of coordinated, internally programmed series of events affected by the set of gene products.
Apoptosis is a pathway of cell death that is induced by a tightly regulated suicide program in which cells destined to die activate enzymes that degrade the cells' own nuclear DNA and nuclear and cytoplasmic proteins.
Types of apoptosis:
A) Physiological:
1) The programmed destruction of cells during embryogenesis,
-- including implantation, organogenesis, developmental involution, and metamorphosis.
2) Hormone-dependent involution in the adult,
3) Cell deletion in proliferating cell populations
4) Death of host cells that have served their useful purpose
5) Elimination of potentially harmful self-reactive lymphocytes, either before or after their maturation.
6) Cell death induced by cytotoxic T cells.
B) Pathological:
1) Cell death produced by a variety of injurious stimuli, such as radiation and cytotoxic anticancer drugs.
2) Cell injury in certain viral diseases, such as viral hepatitis.
3) Pathologic atrophy in parenchymal organs after duct obstruction, Ex: occurs in the pancreas, parotid gland, and kidney.
4) Cell death in tumors,
Morphology (On electron microscope):
Biochemical features of Apoptosis:
Mechanism of apoptosis:
1) An initiation phase, during which caspases become catalytically active, and
2) An execution phase, during which these enzyme act to cause cell death.
1) Initiation phase: Initiation of apoptosis occurs by the signal from two distinct but convergent pathways-
Both pathways converge to activate caspases.
2) Executation phase: mediated by proteolytic cascade. Execution caspases activate cytoplasmic endonucleases and proteases thus degrade nuclear and cytoskeletal proteins, resulting in fragmentation of nuclear chromatin and breakdown of the cytoskeletal proteins.
The end result is the formation of apoptotic bodies containing intracellular organelles and other cytosolic components; which is uptake by phagocytic cells.
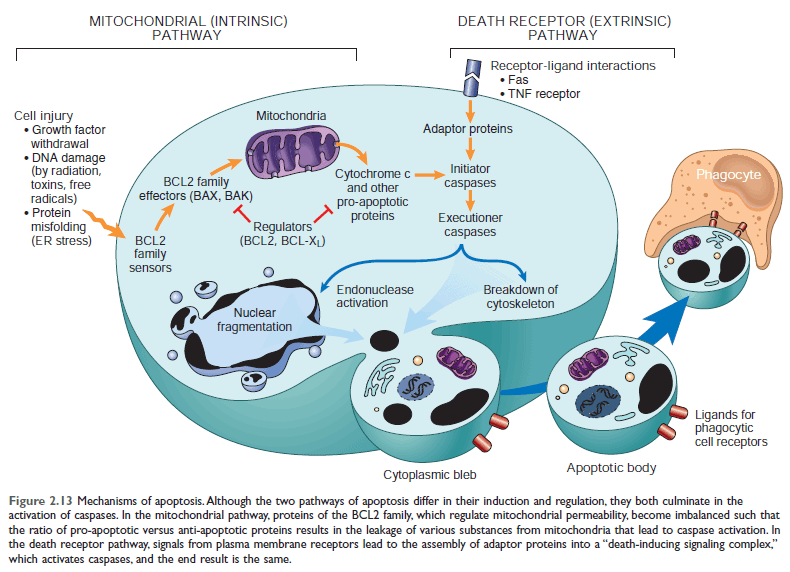
Figure: Mechanism of apoptosis (Page 44) 10th Edition
| Feature | Necrosis | Apoptosis |
| Cell size | Enlarged (swelling) | Reduced (shrinkage) |
| Nucleus | Pyknosis → karyorrhexis → karyolysis | Fragmentation into nucleosome-size fragments |
| Plasma membrane | Disrupted | Intact; |
| Cellular contents | Enzymatic digestion; may leak out of the cell | Intact; may be released in apoptotic bodies |
| Adjacent inflammation | Frequent | No |
| Physiologic or pathologic role | Invariably pathologic | Often physiologic, means of eliminating unwanted cells; may be pathologic after some forms of cell injury, especially DNA damage |
Ref 10th Edition
Note: nuclear condensation (pyknosis), followed by fragmentation (karyorrhexis) and dissolution of the nucleus (karyolysis)
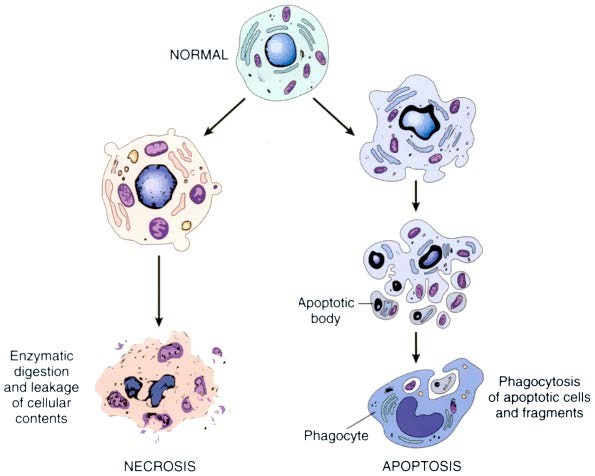
FIGURE: The sequential ultrastructural changes seen in necrosis (left) and apoptosis (right). In apoptosis, the initial changes consist of nuclear chromatin condensation and fragmentation, followed by cytoplasmic budding and phagocytosis of the extruded apoptotic bodies. (7th Edition)
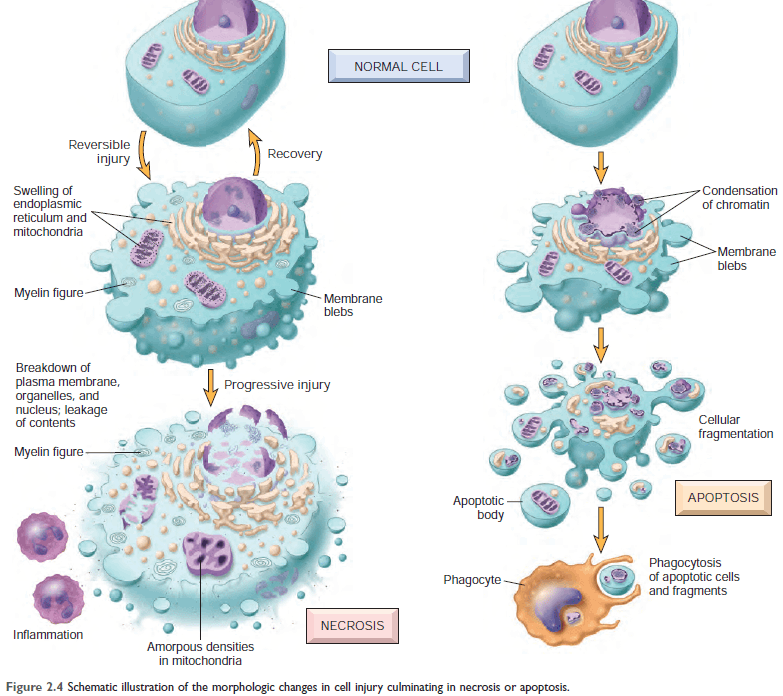
Figure: Morphologic changes in necrosis & apoptosis [Ref Robbin's 10th Edition (Page 38)]
Intracellular accumulations:
Intracellular accumulation is the accumulation of abnormal amounts of various substances due to manifestations of metabolic derangement in the cell.
Categories:
1. A normal cellular constituent: Water, Lipids, proteins, carbohydrates
2. An abnormal substance:
3. Pigment.
Site: cytoplasm, within organelles (typically lysosomes), or in the nucleus.
Source: substances that are synthesized by the affected cells or are produced elsewhere.
Mechanisms: There are four main mechanisms
1. Inadequate removal of normal substance (secondary to defects in mechanisms of packaging and transport eg, fatty change).
2. Accumulation of an abnormal endogenous substance (as a result of defects in its folding, packaging, transport, or secretion eg; a mutated form of α1 antitrypsin)
3. Failure to degrade a metabolite due to inherited enzyme deficiencies (eg: storage diseases)
4. Deposition and accumulation of an abnormal exogenous substance (eg: accumulation of carbon and silica particles).
Importance:
Accumulation of Lipids:
All major classes of lipids can accumulate in cells:
Accumulation of Protein
PROTEINS
1. Russell bodies
2. Accumulation of cytoskeletal proteins is divided into five classes –
Alcoholic hyaline is an eosinophilic cytoplasmic inclusion in liver cells that is characteristic of alcoholic liver disease and is composed predominantly of keratin intermediate filaments
The neurofibrillary tangle found in the brain in Alzheimer's disease contains neurofilaments and other proteins
Accumulation of Pigments.
Exogenous Pigments:
The most common exogenous pigment is a carbon (coal dust),
Example
Endogenous Pigments
Lipofuscin, also known as lipochrome or wear-and-tear pigment. The term is derived from the Latin (fuscus, brown), referring to brown lipid.
Melanin, derived from the Greek (melas, black), is a brown-black pigment.
Hemosiderin is a hemoglobin-derived, golden yellow-to-brown, granular or crystalline pigment.
Pathologic Calcification
Pathological calcification is abnormal tissue deposition of calcium salts together with small amounts of iron, magnesium, and other mineral salts.
Types of pathological calcification:
Dystrophic calcification:
When deposition occurs in dead or dying tissue, despite normal serum calcium levels, known as dystrophic calcification.
Sites of Dystrophic calcification::
Metastatic calcification:
Metastatic calcification occurs in normal tissue when there is hypercalcemia.
Sites of metastatic calcification: May occur widely throughout the body but principally:
Causes of hypercalcemia:
Pathogenesis:
Calcium ions are deposited in the form of hydroxyapatite.
The process has two major phases:
(Both can occur intracellularly and extracellularly.)
Initiation:
Initiation of intracellular calcification occurs in the mitochondria of dead or dying tissue. Initiation of extracellular calcifications occurs in membrane-bound vesicles derived from degenerating or aging cells.
Propagation:
Propagation of crystal formation depends on the concentration of Calcium and PO4.
Importance of calcification:
Comments (0)