Amino Acid/Proteins & their metabolism [Part 2] (Viva)
6 years ago 6036
![Amino Acid/Proteins & their metabolism [Part 2] (Viva)](https://emedicodiary.com/images/queimg/e48dd3269103755bcea81e3bb390a96f.jpg)
Q.109 What is transamination?
Transamination is an intermolecular transfer of an amino group of an amino acid to a keto group of keto acid resulting in the formation of corresponding keto acid and amino acid.
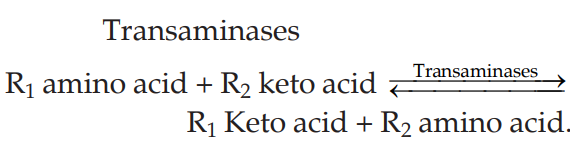
Q.110 What is the co-factor required in transamination?
Pyridoxal phosphate
Q.111 Name the tissues in which transamination reactions occur.
Transamination takes place principally in
Q.112 What are the amino acids which participate most in transamination?
Alanine, aspartic acid, glutamic acid
Q.113 What are the amino acids which does not participate in transamination?
Lysine, threonine.
Q.114 Name two specific transaminases which are of clinical importance. What are the substrates and end-products?
Aspartate transaminase (or aspartate aminotransferase): Previously used to be called as “S-GOT”— serum glutamate oxaloacetate transaminase.
Asparate (Donor amino acid) + α-oxoglutrate (recipient keto acid) — > OAA (new ketoacid) + Glutamate (new amino acid)
Alanine transaminase (or Alanine aminotransferase): Previously used to be called as “S-GPT”— serum glutamate “pyruvate transaminase”.
Alanine (Donor amino acid) + α-oxoglutrate (recipient keto acid) — > Pyruvic acid (new ketoacid) + Glutamate (new amino acid)
Note: In both cases, glutamic acid is produced as new amino acid.
Q.115 What are normal levels of S-GOT and S-GPT?
Q.116 What is deamination? What are the types?
Deamination is the process by which N of amino acid is removed as NH3 2 types of deamination:
Q.117 What is the site of oxidative deamination?
Q.118 What is the nature of enzyme and coenzyme that take part in oxidative deamination?
Enzyme: L-amino acid oxidase which acts specifically on L-amino acids and oxidatively deaminates to form NH3.
Coenzyme: is flavoprotein (FMN) which acts as H-acceptor.
Nature of the enzyme: The L-amino acid oxidases are auto-oxidizable flavoproteins. The reduced flavoproteins FMN. H2 is re-oxidized at the substrate level directly by molecular O2 forming H2O2 which is converted to H2O by the enzyme catalase.
Q.119 What are the toxic substances that can form in the oxidative deamination of Lamino acids by “L-amino acid oxidase?
Q.120 What is the function of the presence of D-amino acid oxidase in cells, when it is known there is an absence of D-amino acids in cells?
There are no D-amino acids in the body tissues, hence it is not clear about the presence of D-amino acid oxidase in cells and its functions. Probably the dietary deamino acids may be oxidized by D-amino acid oxidase and be isomerized through their ketoacids to L-amino acids.
Q.121 Does oxidative deamination fulfill a major role in the formation of NH3?
L-amino acid oxidase does not fulfill a major role in mammalian amino acid catabolism and formation of NH3 as
Q.122 What is non-oxidative deamination? Give two examples.
Certain amino acids like OH-amino acids, histidine, and S-containing amino acids can be deaminated non-oxidatively by specific enzymes to form NH3. They also do not fulfill a major role in NH3 formation.
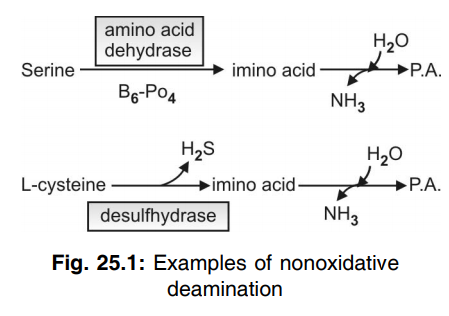
Q.123 State the principal method by which NH3 is formed from L-amino acids.
Q.124 List the sources of NH3 in the body.
Q.125 How albumin and globulin are separated?
Albumin can be separated by full saturation of ammonium sulfate. Globulin can be separated by half-saturation of ammonium sulfate.
Q.126 Name some biologically active peptides.
Q.127 Which amino acid gives rise to thyroxine?
Phenylalanine
Q.128 What is the fate of ammonia?
Q.129 Describe the urea cycle. What is another name for the urea cycle?
This is a cycle through which the toxic NH3 in the body is detoxicated to form non-toxic urea in the liver. An alternative name for the urea cycle is Krebs’ Henseleit cycle. In this process, one molecule of NH3 and the molecule of CO2 are converted into urea through five sequential enzymatic reactions in each turn of the cycle. Ornithine is regenerated in the last reaction which acts as a catalytic agent and repeats the cycle again.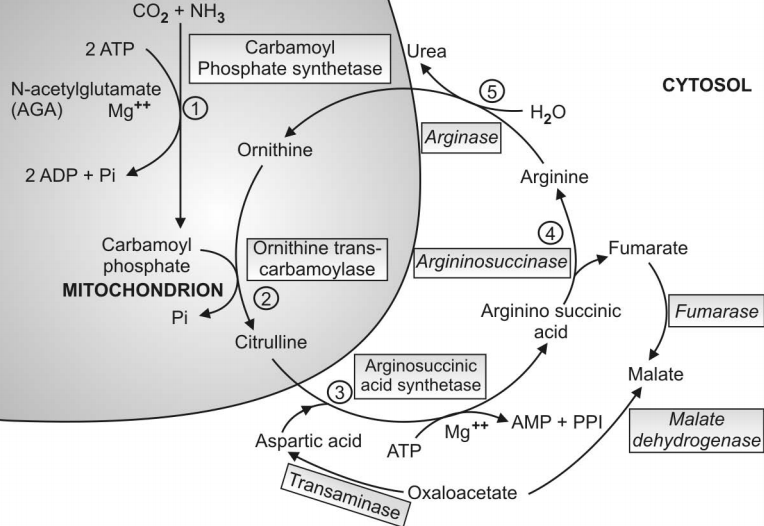
Q.130 What are non-protein nitrogenous substances?
Nitrogen-containing substances other than amino acids or proteins are called nonprotein nitrogenous substances. They are urea, uric acid, creatine, purine, pyrimidine, amino acids, ammonia, etc.
Q.131 Which organ is associated with urea formation?
Liver is the only organ that can form urea and all the enzymes involved have been isolated from the liver tissue.
Q.132 Can other organs like the kidney and brain synthesize urea?
Kidney:
Can form up to arginine but cannot form urea as enzyme “arginase” is absent in kidney tissues.
Brain:
Can synthesize urea if citrulline is available but lacks the enzyme ornithine transcarbamoylase which forms citrulline from ornithine.
Q.133 State the five steps of the urea cycle indicating the enzymes involved and the location of the enzyme.
| Enzyme | Location | ||
| Reaction 1 : | Synthesis of carbamoyl-P | Carbamoyl synthetase I | Mitochondria |
| Reaction 2 : | Synthesis of citrulline | Ornithine transcarbamoylase | Mitochondria |
| Reaction 3 : | Synthesis of Arginino- succinate | Arginino- succinate synthetase | Cytosol |
| Reaction 4 : | Cleavage of arginino- succinate | Arginino-succinase | Cytosol |
| Reaction 5 : | Cleavage of arginine to form urea and ornithine | Arginase | Cytosol |
Q.134 What is the role of N-acetyl glutamate (AGA) in the first reaction in the formation of carbamoyl-P?
N-acetyl glutamate (AGA) acts as an allosteric activator of the enzyme carbamoyl synthetase I, it brings about conformational changes in the enzyme so that the second molecule of ATP can bind.
Q.135 Show schematically the first reaction, synthesis of carbamoyl-P. What are the starting materials? Is biotin necessary?
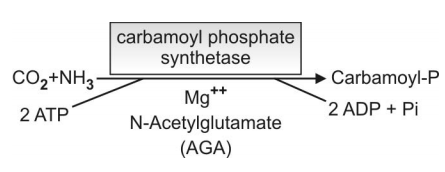
Q.136 How many amino acids take part in the urea cycle?
5 amino acids participate in the urea cycle.
They are:
Q.137 What is the energy requirement in the formation of one molecule of urea in the urea cycle?
3 molecules of ATP are utilized for the formation of one molecule of urea. As one ATP is converted to AMP, it is equivalent to utilizing 4 high energy bonds.
Q.138 Urea contains two nitrogen (NH2 groups), what are the sources of the two nitrogens?
Q.139 What is the normal blood level of urea in the body?
Q.140 In what conditions the blood level of urea increase:
Causes may be:
Q.141 Mention some prerenal causes.
Conditions in which plasma volume/body fluid are reduced, e.g.:
Q.142 State some renal causes:
Q.143 Mention some post-renal causes.
Q.144 What is glutamine?
Glutamine is chemically δ-amide of αamino glutaric acid. Glutamine formation helps in the removal of NH3 which is toxic (detoxication of NH3).
Glutamine serves as an important reservoir of nitrogen in tissues that is readily available and can be drawn upon for various synthetic processes when the body needs.
Q.145 How glutamine is synthesized in the body? Name the tissues involved in the synthesis of glutamine.
Glutamine is synthesized in tissues like:
It is formed from:
– Glutamic acid and
– NH3 by the action of a mitochondrial enzyme “Glutamine synthetase” in presence of ATP and Mg++.
The reaction is “irreversible”, so that glutamine cannot reform glutamic acid and NH3 by the same enzyme.
Q.146 How NH3 can be obtained from glutamine?
Glutamine is a reservoir of NH3 and a readily available source of NH3.
Glutamine is hydrolyzed readily by a specific enzyme glutaminase. The reaction is ‘irreversible’ and can occur in various tissues like the liver, kidney, brain, and retina.
Q.147 State some important functions of glutamine in the body.
Q.148 What are the clinical manifestations of ammonia intoxication? The symptoms of NH3 intoxication include:
Q.149 How much urea is excreted daily in urine?
20 to 30 gm in 24 hours urine.
Q.150 Name the inherited disorders associated with urea cycle.
| Hyperammonemia type I—due to deficiency of enzyme carbamyl-P-synthetase I |
| Hyperammonemia type II, (also called ornithinaemia)—due to the deficiency of enzyme ornithine transcarbamoylase |
| Citrullinemia—due to deficiency of the enzyme argininosuccinate synthetase |
| Argininosuccinic aciduria—due to deficiency of enzyme argininosuccinase |
| Hyperargininemia—due to deficiency of enzyme arginase. |
Q.151 What are the amino acids involved in creatine synthesis?
Glycine, arginine, methionine (as S-adenosyl methionine).
Q.152 What is the role of creatine phosphate?
As a source of energy in the contraction of muscles.
Q.153 What are the uses of glycine?
Glycine is involved in the synthesis of
Q.154 Which is the major non-protein nitrogenous constituent of the urine?
Urea
Q.155 Where carbamoyl phosphate synthase II is required?
In pyrimidine de novo biosynthesis
Q.156 What is the role of N-acetyl glutamic acid in the first step of the urea cycle?
N-acetyl glutamic acid is the modifier of the enzyme carbamoyl phosphate synthetase. It keeps the enzyme in the correct information.
Q.157 How much protein is potentially glucogenic?
It has been shown by experimental studies that 60 gm (approximately 58 gm) of glucose are formed and excreted in urine for 100 gm of proteins metabolized. Thus 60% of proteins (amino acids) are potentially glucogenic.
Q.158 Name at least ten amino acids which are glucogenic.
Glycine, alanine, serine, cysteine/cystine, threonine methionine, proline, valine, arginine, glutamate.
Q.159 Name the only amino acid which is ketogenic.
L-leucine
Q.160 Name at least three amino acids which are both glucogenic and ketogenic.
Phenylalanine/tyrosine, tryptophan, isoleucine.
Q.161 1. What is GABA?
2. How it is formed?
3 What is the coenzyme of this reaction?
1. GABA is γ−aminobutyric acid, important in brain metabolism.
2. It is formed by the decarboxylation of glutamic acid.
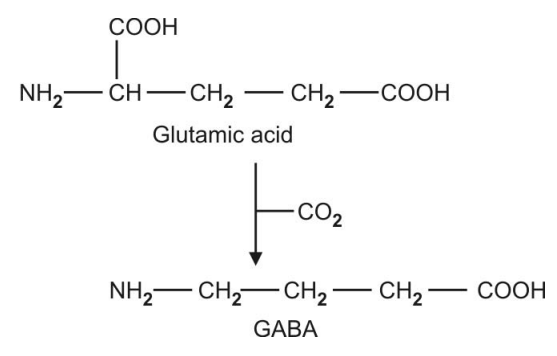
3 .Coenzyme of the reaction is pyridoxal phosphate.
Q.162 Define albinism.
Deficiency of enzyme tyrosinase in the tyrosine metabolism leads to an inborn error of metabolism called albinism.
Q.163 What is tyrosinosis?
Deficiency of enzyme p-hydroxyphenyl pyruvate hydroxylase in tyrosine metabolism leads to tyrosinosis.
Q.164 What is alkaptonuria?
Deficiency of enzyme homogentisic acid oxidase in tyrosine metabolism leads to alkaptonuria.
Q.165 What is phenylketonuria?
Deficiency of the enzyme phenylalanine-4 hydroxylase in tyrosine metabolism leads to phenylketonuria.
Q.166 Name some biological important amines.
Q.167 What are catecholamines?
Catecholamines are
Q.168 Name at least five biogenic amines of clinical importance. State the amino acid from which it is derived and mention biological importance in the body.
| Biogenic amine | Name of the amino acid | Biologic importance |
| Histamine | Histidine | Vasodilator, BP↓ ↑ HCl Pepsin-, Liberated in anaphylactic reaction |
| γ-aminobutyric acid (GABA) | Glutamic acid | Presynaptic inhibitor in brain Forms a by-pass in the TCA cycle (GABA shunt) |
| Taurine | Cysteic acid (derived from cysteine) | Constituent of bile acid (taurocholic acid) |
| Tryptamine | Tryptophan | Tissue hormone, a derivative of 5-OH tryptamine (serotonin) Vasoconstriction, BP↑ |
| Ethanol-amine | Serine | Forms choline Constituent of PL like cephalin. |
Q.169 What is GABA shunt? Show schematically.
GABA by its conversion to succinic acid can form a “by-pass” in the TCA cycle and this is called as GABA shunt
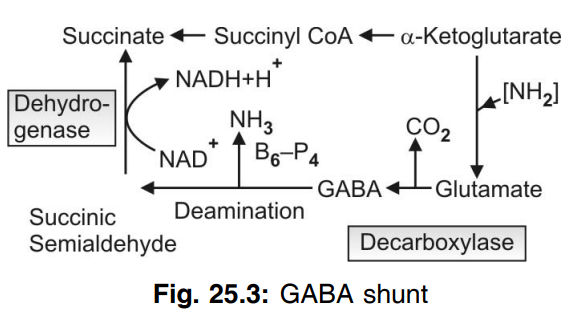
Q.170 Name the polyamines.
Q.171 What is carbamoyl-P synthetase II? What is its function?
Carbamoyl-P-synthetase II is an enzyme present in the cytosol. It can form carbamoyl-P from CO2 and amide-N of glutamine, which is used in pyrimidine synthesis.
Q.172 What are the aromatic amino acids? Name them, which is essential and which is not.
Q.173 State how phenylalanine is converted to tyrosine in the body.
Reactions occur in two stages:
Stage 1:
Reduction of molecular O2 to H2O and conversion of phenylalanine → to tyrosine. Reduced from of folic acid F.H4 acts as H-donor to molecular O2 and is converted to F.H2. In this stage, there is incorporation of one atom of molecular O2 to p-position of phenylalanine to form tyrosine, while another atom of O2 is reduced to H2O.
Stage 2:
Reduction of F.H2 to reform F.H4 takes place by NADPH, acting as an H-donor, catalyzed by the enzyme dihydrobiopterin reductase.
Note: Both the reactions are irreversible.
Q.174 What is meant by sparing action?
The feeding of tyrosine/or cysteine decreases the need of phenylalanine/or methionine in the diet respectively. This is called “sparing” action. Phenylalanine/or methionine is spared from the synthesis of tyrosine/or cysteine respectively.
Q.175 Tyrosine is a non-essential amino acid. In which condition it becomes an essential amino acid?
In phenylketonurias, where phenylalanine cannot be converted to tyrosine in the body due to inherited absence of the enzyme phenylalanine hydroxylase, tyrosine becomes an essential amino acid to the patient.
Q.176 What is albinism?
It is an inherited disorder of tyrosine metabolism in which the enzyme tyrosinase is lacking resulting in non-formation of melanin pigment for which the hair, skin, and retina of the eye are not black.
Q.177 What is the relationship between cysteine and cystine?
Q.178 What is active methionine? What is it known as chemically?
“Active methionine” is an active compound in which the CH3 group is biologically labile and it acts as a methyl (CH3) group donor to a suitable “acceptor” so that it can form biologically important compounds.
Chemically “active methionine” is S-adenosyl methionine.
Q.179 What is meant by transmethylation reaction?
Certain compounds of the body, with structures containing CH3 group attached to an atom other than carbon can take part in enzymic reactions, whereby these CH3 groups are transferred to a suitable “acceptor” which has no methyl group. Such reactions are called a transmethylation reaction.
Q.180 Name the compounds which possess a biologically labile methyl group.
Active methionine (S-adenosyl methionine): containing -S ~ CH3 group.
Choline: containing -CH3 groups attached to N as follows:
CH5
N+ — CH5
CH5
Betaine: an oxidation product of choline.
Q.181 Give Five examples of transmethylation reactions, where active methionine acts as a methyl donor.
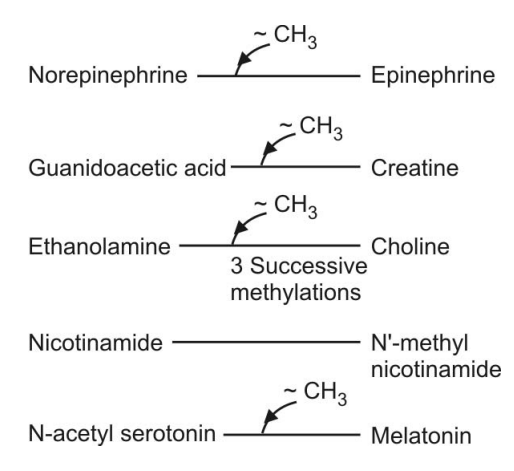
Q.182 Enumerate the metabolic role or biomedical importance of L-methionine.
Q.184 What is homocystinuria? What is the enzyme deficiency?
An inherited disorder of metabolism of L-methionine or more specifically its metabolic intermediates homocysteine/ or homocystine.
Enzyme deficiency: Cystathionine synthetase enzyme deficiency leads to the accumulation of homocystine which is excreted in urine.
Q.185 State five metabolic role/or biomedical importance of glutathione in the body.
Q.186 What is serotonin?
Serotonin is chemically 5-OH tryptamine (5- HT), and it is formed from amino acid tryptophan. It is present in blood and is produced in tissues like gastric mucosa, intestine, brain, mast cells, and platelets. (Probably stored in Platelets?)
Q.187 How serotonin is synthesized by the serotonin-producing cells in the body?
Tryptophan is first hydroxylated to form 5-OH tryptophan in the liver by hydroxylase enzyme in presence of O2 and NADPH.
5-OH tryptophan is next decarboxylated by the enzyme decarboxylase to form 5-OH tryptamine (5-HT), called serotonin, the reaction requires B6P as a coenzyme.
Q.188 Name the enzyme which degrades serotonin. What is the degradation product?
Enzyme is monoamine oxidase (MAO)
Degradation product is 5-OH-Indole acetic acid (5-HIAA) which is excreted in urine.
Q.189 In which condition excessive amount of serotonin is produced in the body?
Q.190 What is melatonin?
Chemically melatonin is “N-acetyl-5- methoxy serotonin”. It is a hormone produced by the pineal gland and peripheral nerves of man and some other higher animals.
The hormone lightens the color of melanocytes in the skin of the frog and blocks the action of MSH (Melanocyte stimulating hormone) and ACTH.
Q.191 What is Hartnup’s disease?
It is an inherited disorder associated with the metabolism of tryptophan. The clinical symptoms are mental retardation, intermittent cerebellar ataxia, and skin rash.
Urine of patients with Harnup’s disease contains increased amounts of indoleacetic acid and tryptophan.
Q.192 What is the metabolic defect in Hartnup’s disease?
An inherited deficiency of the enzyme tryptophan dioxygenase in the liver.
Could also be due to a defective indole transport and consequent impairment of tryptophan metabolism.
Q.193 What is the relationship between creatine-P and creatinine?
Q.194 Show schematically the biosynthesis of creatine.
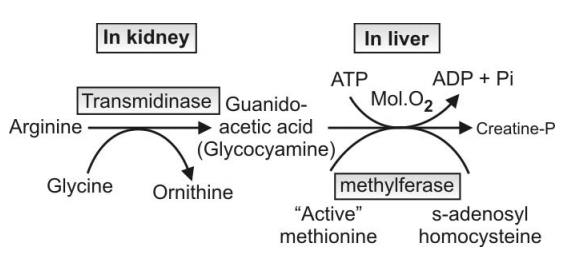
Q.195 Where does the formation of creatinine occur?
In Liver and in Kidney.
Q.196 What is meant by the creatinine clearance test.
Endogenous creatinine clearance is used as a renal function test. At normal levels of creatinine in the blood, this metabolite is filtered at the glomerulus but neither secreted nor reabsorbed by the tubules. Hence its clearance measures the glomerular filtration rate (GFR).
Q.197 What is the normal endogenous clearance?
Normal values for creatinine clearance varies from 95 to 105 ml/min.
Q.198 What are the biological importance and metabolic role of arginine?
Also read: Amino Acid/Proteins & their metabolism [Part 1] (Viva)
Comments (0)